Infusions every 8 weeks after 3 induction doses. REMICADE® is administered by intravenous (IV) infusion over a period of not less than 2 hours.1
5 mg/kg IV given at 0, 2, and 6 weeks
as an induction regimen
5 mg/kg IV given every 8 weeks thereafter
as a maintenance regimen
Dosage: 5 mg/kg IV
Patient’s Weight:
*Total dose in mg = 5 mg/kg dose x kg.
†Vials = total dosage/100 (rounded up to nearest whole vial).
‡mL reconstituted REMICADE® = total dosage/10 (rounded to nearest tenth).
Calculation based on recommended 5 mg/kg dosage.
Enter the patient’s weight in pounds (pounds are automatically converted to kilograms). Then hit Enter, and the appropriate dosage amount and number of single vials will populate. Remember to discard any unused solution remaining in the vials.
The recommended dosage of REMICADE® is 5 mg/kg given as an intravenous induction regimen at 0, 2, and 6 weeks, followed by a maintenance regimen of 5 mg/kg every 8 weeks thereafter for the treatment of active PsA. REMICADE® can be used with or without methotrexate.
Before infusing REMICADE®, provide patients or their caregivers with the Medication Guide for REMICADE®. You can obtain a printable PDF of the full Prescribing Information and the Medication Guide here. Refer to the Preparation and Administration Instructions and important information on what to do in the event your patient experiences an infusion reaction.
REMICADE® is intended for use under the guidance and supervision of a healthcare provider. The supplied lyophilized powder must be reconstituted and diluted prior to administration. The infusion solution should be prepared and administered by a trained medical professional using aseptic technique by the following procedure:

Calculate the dose, total volume of reconstituted REMICADE® solution required, and the number of REMICADE® vials needed. More than one vial may be needed for a full dose.
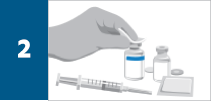
Reconstitute each 100-mg REMICADE® vial with 10 mL of Sterile Water for Injection, USP, to obtain a concentration of 10 mg/mL, using a syringe equipped with a 21-gauge or smaller needle as follows:
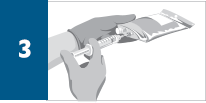
Dilute the total volume of the reconstituted REMICADE® solution to 250 mL* with sterile 0.9% Sodium Chloride Injection, USP, (do not dilute with any other diluent) as follows:
*For volumes greater than 250 mL, either use a larger infusion bag (eg, 500 mL) or multiple 250-mL infusion bags to ensure that the concentration of the infusion solution does not exceed 4 mg/mL.
The REMICADE® infusion should begin within 3 hours of reconstitution and dilution. The infusion must be administered intravenously for at least 2 hours with an infusion set with an in-line, sterile, non-pyrogenic, low-protein-binding filter (pore size of 1.2 µm or less).

Given that the vials do not contain antibacterial preservatives, discard any unused portion of the infusion solution (do not store for reuse).
No physical biochemical compatibility studies have been conducted to evaluate the co-administration of REMICADE® with other agents. REMICADE® should not be infused concomitantly in the same intravenous line with other agents.
Infusion reactions may occur with numerous IV medications, including REMICADE®. In clinical trials of REMICADE®, the majority of infusion reactions were mild to moderate. Most of these reactions were manageable and responded to appropriate treatment steps.
Serious cerebrovascular accidents, myocardial ischemia/infarction (some fatal), hypotension, hypertension, and arrhythmias have been reported during and within 24 hours of initiation of REMICADE® infusion. Cases of transient visual loss have been reported during or within 2 hours of infusion of REMICADE®. Monitor patients during infusion and if serious reaction occurs, discontinue infusion. Further management of reactions should be dictated by signs and symptoms.
Thorough patient assessment and screening for hypersensitivity reactions are key to helping to prevent infusion-related events.
Prior to treatment, ensure appropriate personnel and medication are available to treat reactions (eg, hypersensitivity, other reactions) that occur during infusion and shortly after infusion. Prior to infusion with REMICADE®, patients may be premedicated with histamine-1 receptor antagonists, histamine-2 receptor antagonists, acetaminophen, and/or corticosteroids.
For mild to moderate reactions during the infusion, consider slowing or stopping the infusion. Upon resolution of these reactions, may reinitiate at a lower infusion rate and/or with histamine-1 receptor antagonists, histamine-2 receptor antagonists, acetaminophen, and/or corticosteroids. Discontinue the infusion if the mild to moderate reactions reoccur.
Discontinue the infusion if severe hypersensitivity reactions occur during the infusion.
Reference: 1. REMICADE® [Prescribing Information]. Horsham, PA: Janssen Biotech, Inc.
Targan et al conducted a multicenter, randomized, double-blind trial in 108 patients with moderately to severely active CD (baseline CDAI ≥220 and ≤400) unresponsive to conventional therapy. Patients were randomized to receive a single infusion of placebo (n=25) or REMICADE® 5 mg/kg IV (n=27), 10 mg/kg IV (n=28), or 20 mg/kg IV (n=28). The primary endpoint was clinical response at Week 4.5
Note: The recommended dose of REMICADE® is 5 mg/kg given as an IV induction regimen at 0, 2, and 6 weeks followed by a maintenance regimen of 5 mg/kg IV every 8 weeks thereafter for the treatment of adults with moderately to severely active Crohn’s disease.1
ACCENT I is a 1-year, multicenter, randomized, double-blind trial of REMICADE® in 545 patients with moderately to severely active CD (CDAI ≥220 and ≤400). All patients received an initial dose of REMICADE® 5 mg/kg IV. Patients were then randomized based on clinical response at Week 2 to 1 of 3 treatment groups through Week 541,6:
The coprimary endpoints of the trial were the proportion of patients responding at Week 2 who were in remission at Week 30 and time to loss of response through Week 54.1,6
Note: The recommended dose of REMICADE® is 5 mg/kg given as an IV induction regimen at 0, 2, and 6 weeks followed by a maintenance regimen of 5 mg/kg IV every 8 weeks thereafter for the treatment of adults with moderately to severely active Crohn’s disease.1
Results from SONIC, a multicenter, randomized, double-blind, controlled, phase IIIb trial of 508 patients with moderately to severely active CD (baseline CDAI ≥220 and ≤450). At baseline, mean CDAI score for all patients was 287; median duration of CD ranged from 2.2 to 2.4 years. Patients were randomly assigned to 1 of 3 treatment groups through Week 30. In the primary study phase, patients received REMICADE® 5 mg/kg or placebo infusions at Weeks 0, 2, 6, 14, and 22; patients also received AZA capsules at a dose of 2.5 mg/kg/day or placebo capsules daily through Week 30. Patients completing treatment through Week 30 (main study) were eligible to enter the study extension if, in the opinion of the investigator, the patient could benefit from continued treatment. Patients eligible for participation in the extension (N=280) continued to receive their initial treatment through Week 50. The Week-50 analysis included patients who did not enter the study extension. These patients were assumed nonresponders and not to be in steroid-free remission at Week 50. The primary endpoint of the study was the proportion of patients in corticosteroid-free remission at Week 26. Secondary endpoints included mucosal healing at Week 26 and corticosteroid-free remission at Week 50. Monitoring for adverse events was performed through Week 54.
REACH (A Randomized, multicenter, open-label study to Evaluate the safety and efficacy of Anti-TNF-α Chimeric monoclonal antibody in pediatric subjects with moderate to severe Crohn's disease) was a controlled trial that evaluated the safety and efficacy of REMICADE® in 112 pediatric patients aged 6 to 17 years with moderately to severely active CD. All patients received induction dosing of 5 mg/kg IV REMICADE® at Weeks 0, 2, and 6. At Week 10, 103 patients were randomized to a maintenance regimen of REMICADE® 5 mg/kg IV given either every 8 weeks or every 12 weeks. Induction of clinical response, the primary endpoint, was evaluated 10 weeks after the 3-dose induction regimen (Weeks 0, 2, and 6). At Week 54, clinical response, clinical remission, and change from baseline in average daily corticosteroid use were evaluated as secondary efficacy endpoints.5
Note: REMICADE® 5 mg/kg IV every 12 weeks is not an approved maintenance dose for REMICADE®.1
The safety and efficacy of REMICADE® were evaluated in the Active Ulcerative Colitis Trial (ACT 1) (N=364), a randomized, double-blind, placebo-controlled, multicenter trial conducted in patients with moderately to severely active ulcerative colitis who had an inadequate response or were intolerant to conventional therapy. Patients presented with a Mayo score between 6 and 12, and an endoscopy subscore of ≥2. Prior failed or intolerable therapies in ACT 1 included oral corticosteroids, 6-mercaptopurine (6-MP), or azathioprine (AZA). Patients were randomized to the following treatment groups: REMICADE® 5 mg/kg IV (n=121), REMICADE® 10 mg/kg IV (n=122), or placebo (n=121). Infusions were administered at Weeks 0, 2, and 6, and every 8 weeks thereafter through Week 46.5
Final efficacy evaluations were completed 8 weeks following the last infusion. Concomitant treatment with stable doses of aminosalicylates, corticosteroids, and/or immunomodulators was permitted throughout the study. Of patients receiving steroids at baseline, tapering was allowed beginning at Week 8. The primary efficacy endpoint was clinical response at Week 8; secondary endpoints included clinical remission and mucosal healing.5
Note: The recommended dose of REMICADE® is 5 mg/kg given as an IV induction regimen at 0, 2 and 6 weeks followed by a maintenance regimen of 5 mg/kg IV every 8 weeks thereafter for the treatment of adult patients with moderately to severely active ulcerative colitis.1
The safety and efficacy of REMICADE® were evaluated in ACT 2 (N=364) (ACT=Active Ulcerative Colitis Trial), a randomized, double-blind, placebo-controlled, multicenter trial conducted in patients with moderately to severely active ulcerative colitis who had an inadequate response or were intolerant to conventional therapy. Patients presented with a Mayo score between 6 and 12, and an endoscopy subscore of ≥2. Prior failed or intolerable therapies in ACT 2 included oral corticosteroids, 6-mercaptopurine (6-MP), or azathioprine (AZA) and/or aminosalicylates. Patients were randomized to the following treatment groups: REMICADE® 5 mg/kg IV (n=121), REMICADE® 10 mg/kg IV (n=120), or placebo (n=123). Infusions were administered at Weeks 0, 2, and 6, and every 8 weeks thereafter through Week 22.5
Final efficacy evaluations were completed 8 weeks following the last infusion. Concomitant treatment with stable doses of aminosalicylates, corticosteroids, and/or immunomodulators was permitted throughout the study. Of patients receiving steroids at baseline, tapering was allowed beginning at Week 8. The primary efficacy endpoint was clinical response at Week 8; secondary endpoints included clinical remission and mucosal healing.5
Note: The recommended dose of REMICADE® is 5 mg/kg given as an IV induction regimen at 0, 2 and 6 weeks followed by a maintenance regimen of 5 mg/kg IV every 8 weeks thereafter for the treatment of adult patients with moderately to severely active ulcerative colitis.1
The REMICADE® Pediatric Ulcerative Colitis (UC) trial was a multicenter, phase 3, randomized, open-label, parallel-group trial to evaluate the safety and efficacy of REMICADE® in pediatric patients aged 6 to 17 years with moderately to severely active UC (N=60; Mayo score of 6 to 12; endoscopic subscore ≥2) and an inadequate response to conventional therapies. The primary objectives of the study were to evaluate clinical response after a 3-dose induction regimen of REMICADE® 5 mg/kg IV and the safety of REMICADE® during induction and maintenance regimens. Secondary objectives included the evaluation of 2 REMICADE® maintenance dosing regimens (every 8 weeks and every 12 weeks) in maintaining remission, as measured on the Pediatric Ulcerative Colitis Activity Index (PUCAI); the efficacy of a 3-dose regimen of REMICADE® in the induction of clinical remission, as measured by the Mayo score; and the induction of remission, as measured on the PUCAI.1,5
All patients received induction dosing of REMICADE® 5 mg/kg IV at Weeks 0, 2, and 6. Patients who did not respond to REMICADE® at Week 8 received no further REMICADE® and returned for safety follow-up. At Week 8, 45 patients were randomized to a maintenance regimen of REMICADE® 5 mg/kg IV given either every 8 weeks through Week 46 or every 12 weeks through Week 42.1,5
Note: REMICADE® 5 mg/kg IV every 12 weeks is not an FDA-approved maintenance dosing regimen for REMICADE® in the treatment of pediatric patients with moderately to severely active UC.1
EXPRESS evaluated 378 patients with moderate to severe plaque psoriasis. These patients had ≥10% body surface area (BSA) involvement, a Psoriasis Area and Severity Index (PASI) score of ≥12, and were candidates for systemic or phototherapy. Patients were randomized to placebo or REMICADE® (infliximab) at a dose of 5 mg/kg IV at Weeks 0, 2, and 6 (induction therapy), followed by maintenance therapy (5 mg/kg IV) every 8 weeks. At Week 24, the placebo group crossed over to REMICADE® induction (5 mg/kg IV), followed by maintenance therapy (5 mg/kg IV) every 8 weeks. Patients randomized to REMICADE® continued to receive REMICADE® 5 mg/kg IV every 8 weeks through Week 46. The primary endpoint was the proportion of patients achieving ≥75% improvement in PASI from baseline to Week 10.
EXPRESS II evaluated 835 patients with moderate to severe plaque psoriasis. These patients had ≥10% body surface area (BSA) involvement, a Psoriasis Area and Severity Index (PASI) score of ≥12, and were candidates for systemic therapy or phototherapy. Patients were randomized to placebo or REMICADE® (infliximab) at doses of 3 mg/kg IV or 5 mg/kg IV at Weeks 0, 2, and 6 (induction therapy). At Week 14, patients in the REMICADE® arms continued on their original dose but were randomized to either an every 8 week scheduled maintenance therapy or an as needed (PRN) maintenance therapy through Week 46. Patients randomized to the PRN maintenance therapy group received their original REMICADE® dose when baseline improvement in PASI was <75% and received placebo if PASI improvement was ≥75%. Maintenance visits occurred monthly. At Week 16, the placebo group crossed over to REMICADE® induction therapy (5 mg/kg IV), followed by a maintenance therapy every 8 weeks. The primary endpoint was proportion of patients achieving PASI 75 at Week 10.
SPIRIT evaluated 249 patients with plaque psoriasis. These patients had ≥10% body surface area (BSA) involvement, a Psoriasis Area and Severity Index (PASI) score of ≥12, and had previously received either psoralen plus ultraviolet A treatment (PUVA) or other systemic therapy for their psoriasis. Patients were randomized to either placebo or REMICADE® (infliximab) at doses of 3 mg/kg IV or 5 mg/kg IV at Weeks 0, 2, and 6 (induction therapy). At Week 26, patients with a static physician’s global assessment (sPGA) score of moderate or worse (≥3 on a scale of 0 to 5) received an additional dose of the randomized treatment. The primary endpoint was the proportion of patients achieving ≥75% improvement in PASI from baseline at Week 10.
ASSERT (Ankylosing Spondylitis Study for the Evaluation of Recombinant Infliximab Therapy), a 24-week, multicenter, double-blind, placebo-controlled, randomized, phase 3 study in 279 adult patients with active ankylosing spondylitis according to the modified New York criteria for ≥3 months, with Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) ≥4, and spinal pain assessment score ≥4 on a Visual Analog Scale (VAS), each on a scale of 0-10. The primary endpoint was the proportion of patients with a 20% improvement response according to the criteria of the ASAS International Working Group. Patients were randomized to 1 of 2 treatment groups in a 3:8 ratio: placebo (n=78) or REMICADE® 5 mg/kg (n=201) infused at Weeks 0, 2, 6, 12, and 18. Concurrent stable treatment with nonsteroidal anti-inflammatory drugs (NSAIDs), acetaminophen, and tramadol was permitted during the study. Patients were not permitted to be on any disease-modifying antirheumatic drugs (DMARDs) or systemic corticosteroids.
IMPACT 2 (Induction and Maintenance Psoriatic Arthritis Clinical Trial 2): a randomized, double-blind, placebo-controlled, multicenter, phase 3, parallel-group study of REMICADE® in 200 adult patients with active PsA for at least 6 months who had an inadequate response to disease-modifying antirheumatic drugs (DMARDs) or nonsteroidal anti-inflammatory drugs (NSAIDs). Patients had active articular disease (≥5 swollen and tender joints each), psoriatic target skin lesion (≥2 cm in diameter), and either C-reactive protein (CRP) ≥1.5 mg/dL or morning stiffness lasting ≥45 minutes. Stable methotrexate (MTX) doses of ≤25 mg/week at study entry and stable oral corticosteroid doses equivalent to ≤10 mg/day of prednisone were permitted. During the 24-week, double-blind phase, patients received either REMICADE® 5 mg/kg IV (n=100) or placebo (n=100) at Weeks 0, 2, 6, 14, and 22. At Week 16, placebo patients with <10% improvement in swollen and tender joint counts were switched to active treatment and received REMICADE® 5 mg/kg IV at Weeks 16, 18, 22, 30, 38, and 46. At Week 24, all patients receiving placebo crossed over to active treatment and received REMICADE® 5 mg/kg IV (n=91) at Weeks 24, 26, 30, 38, and 46. Primary endpoints included the proportion of patients with ACR20 response at Week 14 and the change from baseline in total modified van der Heijde-Sharp (vdH-S) score at Week 24. Improvement in Psoriasis Area and Severity Index (PASI) was evaluated in psoriatic arthritis patients with baseline body surface area (BSA) ≥3% (n=87, placebo; n=83, REMICADE®).
START (Safety Trial for Rheumatoid Arthritis with REMICADE® Therapy): a 54-week, randomized, multicenter, double-blind, 3-arm, parallel-group, phase 3 study of the safety of REMICADE® in combination with methotrexate (MTX) in adult patients with moderately to severely active RA. Moderately to severely active RA was defined as ≥6 swollen (out of 66 total) and ≥6 tender joints (out of 68 total) for ≥3 months prior to screening. Patients were receiving MTX for ≥3 months before randomization and at a stable dose (≤25 mg/week) for ≥4 weeks before randomization. Patients could continue receiving other conventional disease-modifying antirheumatic drugs (DMARDs) as long as doses had been stable for ≥4 weeks. Doses of nonsteroidal anti-inflammatory drugs (NSAIDs) and oral corticosteroids must have been stable for ≥4 weeks prior to screening.
The primary objective was to assess the relative risk of serious infection within 22 weeks of initiating therapy with REMICADE® + MTX in subjects matching clinical practice demographics (including severity of disease, background DMARD use, and concomitant disease). Secondary objectives measured the safety and efficacy of dose-escalation regimens in patients with an incomplete response to the initial dose of REMICADE® 3 mg/kg every 8 weeks and the safety of REMICADE® + MTX after 1 year.
Patients (N=1084) were randomized in a 1:1:1 ratio to 1 of 3 treatment groups: placebo infusions through Week 14, followed by REMICADE® 3 mg/kg infusions every 8 weeks through Week 46 (Group 1, n=363); REMICADE® 3 mg/kg infusions every 8 weeks through Week 46, with dose escalation from Week 22 to 46 by 1.5 mg/kg increments, if the patient had an inadequate response (Group 2, n=360); and REMICADE® 10 mg/kg infusions every 8 weeks through Week 46 (Group 3, n=361). At Week 26, Group 2 and Group 3 patients received a placebo infusion in order to maintain treatment blind. The median dose of MTX was 15 mg/week.
ATTRACT (Anti-TNF Trial in Rheumatoid Arthritis with Concomitant Therapy): a 2-year, multicenter, double-blind, placebo-controlled, randomized, phase 3 study of REMICADE® with methotrexate (MTX) in 428 patients with moderately to severely active MTX-refractory established RA (MTX use ≥3 months). Moderately to severely active RA was defined as ≥6 swollen joints (out of 66 total) and ≥6 tender joints (out of 68 total) and ≥2 of the following:
Primary endpoints: reduction of signs and symptoms at 30 weeks, inhibition of structural damage at 54 weeks, and improvement in physical function at 102 weeks. Nearly 50% of patients had advanced disease. Nonsteroidal anti-inflammatory drugs (NSAIDs) and corticosteroids (≤10 mg/day) permitted at stable doses; no other disease-modifying antirheumatic drugs (DMARDs) allowed. There were 4 REMICADE® groups: 3 mg/kg IV q8 weeks + MTX (n=86), 3 mg/kg IV q4 weeks + MTX (n=86), 10 mg/kg IV q8 weeks + MTX (n=87), and 10 mg/kg IV q4 weeks + MTX (n=81); patients randomized to placebo + MTX (n=88).
ASPIRE (Active-controlled Study of Patients Receiving Infliximab for the Treatment of Rheumatoid Arthritis of Early Onset): a 54-week, multicenter, double-blind, active treatment-controlled, randomized, phase 3 study of REMICADE® with methotrexate (MTX) in 1004 MTX-naïve adult patients with moderately to severely active early RA (≥3 months and ≤3 years from date of diagnosis). Moderately to severely active RA was defined as ≥10 swollen joints (out of 66 total) and ≥12 tender joints (out of 68 total) and ≥1 of the following:
Concurrent stable treatment with corticosteroids (equivalent to ≤10 mg prednisone per day) and usual doses of nonsteroidal anti-inflammatory drugs (NSAIDs) were permitted. The coprimary endpoints were reduction of signs and symptoms, inhibition of structural damage, and improvement in physical function from baseline to Week 54. Patients were randomized into 1 of 3 treatment groups in a 5:5:4 ratio: REMICADE® 3 mg/kg IV + MTX (n=359), REMICADE® 6 mg/kg IV + MTX (n=363), and placebo + MTX (n=282). REMICADE® or placebo was infused at Weeks 0, 2, and 6, and every 8 weeks thereafter. MTX was started at 7.5 mg/week and gradually increased to 20 mg/week by Week 8. All patients were to maintain a target MTX dose of 20 mg/week for the duration of the trial, whenever possible.
Infusions every 8 weeks after 3 induction doses. REMICADE® is administered by intravenous (IV) infusion over a period of not less than
5 mg/kg IV given at 0, 2, and 6 weeks
as an induction regimen
5 mg/kg IV given every 8 weeks thereafter
as a maintenance regimen
If response is lost, consider treatment with 10 mg/kg IV1
Patients who do not respond by Week 14 are unlikely to respond, and consideration should be given to discontinuing REMICADE® in these patients1
Infusions every 8 weeks after 3 induction doses. REMICADE® is administered by intravenous (IV) infusion over a period of not less than 2 hours.1
5 mg/kg IV given at 0, 2, and 6 weeks
as an induction regimen
5 mg/kg IV given every 8 weeks thereafter
as a maintenance regimen
Infusions every 8 weeks after 3 induction doses. REMICADE® is administered by intravenous (IV) infusion over a period of not less than
5 mg/kg IV given at 0, 2, and 6 weeks
as an induction regimen
5 mg/kg IV given every 8 weeks thereafter
as a maintenance regimen
Infusions every 8 weeks after 3 induction doses. REMICADE® is administered by intravenous (IV) infusion over a period of not less than
5 mg/kg IV given at 0, 2, and 6 weeks
as an induction regimen
5 mg/kg IV given every 8 weeks thereafter
as a maintenance regimen
Infusions every 8 weeks after 3 induction doses. REMICADE® is administered by intravenous (IV) infusion over a period of not less than
5 mg/kg IV given at 0, 2, and 6 weeks
as an induction regimen
5 mg/kg IV given every 8 weeks thereafter
as a maintenance regimen
Infusions every 8 weeks after 3 induction doses. REMICADE® is administered by intravenous (IV) infusion over a period of not less than
3 mg/kg IV given at 0, 2, and 6 weeks
as an induction regimen
3 mg/kg IV given every 8 weeks thereafter
as a maintenance regimen
REMICADE® should be given in combination with methotrexate.
For patients who have an incomplete response, consideration may be given to adjusting the dose up to 10 mg/kg IV or treating as often as every 4 weeks, bearing in mind that risk of serious infections is increased at higher doses.
Infusions every 8 weeks after 3 induction doses. REMICADE® is administered by intravenous (IV) infusion over a period of not less than
5 mg/kg IV given at 0, 2, and 6 weeks
as an induction regimen
5 mg/kg IV given every 8 weeks thereafter
as a maintenance regimen
REMICADE® can be used with or without methotrexate in active PsA.
Infusions every 6 weeks after 3 induction doses. REMICADE® is administered by intravenous (IV) infusion over a period of not less than 2 hours.1
5 mg/kg IV given at 0, 2, and 6 weeks
as an induction regimen
5 mg/kg IV given every 6 weeks thereafter
as a maintenance regimen
cp-129778v1